



First-principles computational modelling
In this area of Condensed Matter Physics the research activity focuses on the modelization of systems of scientific and technological relevance, starting from the calculation of their ground state electronic structure and of its possible excitations. The use, for the solution of the many-body problem, of advanced computational techniques, based on density functional theory (DFT), allows to precisely evaluate several properties (e.g., magnetic, structural, vibrational, transport, etc) and to predict the behaviour of the considered systems in a wide range of possible external conditions. Besides to contributing significantly to the rationalisation of experimental results, the ab initio modeling represents an invaluable tool to design and optimise new systems for specific target applications.
Currently the main research projects under development are concerned with the following topics:
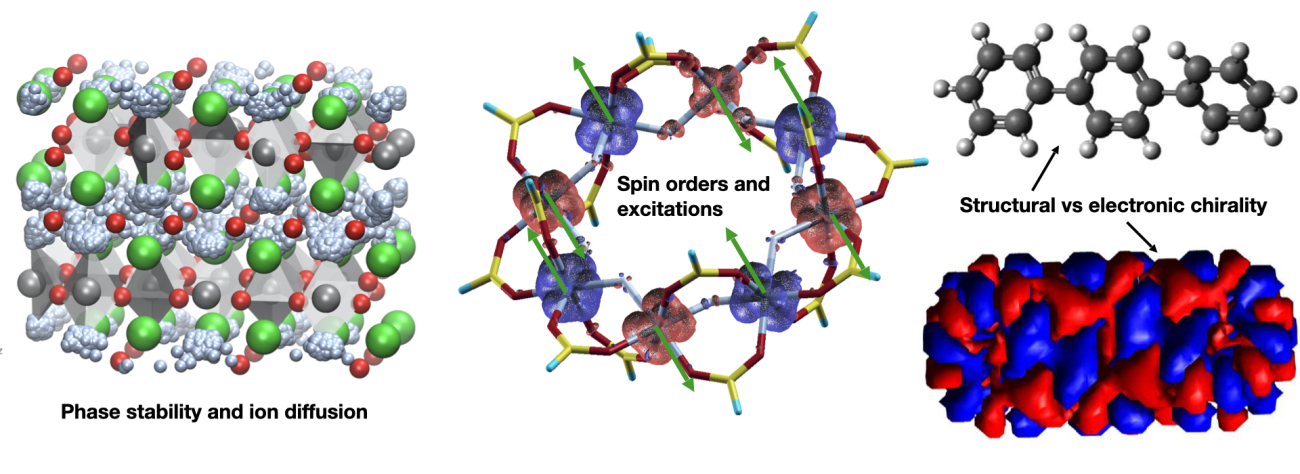
- Phase stability and ionic transport in materials for rechargeable batteries of new generation (in particular, based on Na and F ions)
- Spin configurations and magnetic excitations in molecular magnets (with applications in quantum information, data storage, magneto-refrigeration, etc)
- Inter-relation between structural chirality, spin-polarised transport and magnetic properties in molecules and cristalline systems for the development of applications inherent to spintronics, quantum technologies, chemistry under magnetic control.
The modelling activity is completed by an important theoretical work which, besides improving the comprehension of relevant aspects of the behaviour of most complex systems, aims at enhancing the theoretical foundations and the descriptive and predictive capabilities of DFT by integrating approaches and results from advanced techniques as, for example, quantum field theory or quantum many-body theory and the formalism based on Green’s functions. This theoretical effort is central to address the study of complex systems whose behaviour, characterised by the coupling between different degrees of freedom, results outside the realm of the Fermi-liquid theory.
Principal investigator: Matteo Cococcioni